
Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits
Abstract
Pigeonpea (Cajanus cajan), a tropical grain legume with low input requirements, is expected to continue to have an important role in supplying food and nutritional security in developing countries in Asia, Africa and the tropical Americas. From whole-genome resequencing of 292 Cajanus accessions encompassing breeding lines, landraces and wild species, we characterize genome-wide variation. On the basis of a scan for selective sweeps, we find several genomic regions that were likely targets of domestication and breeding. Using genome-wide association analysis, we identify associations between several candidate genes and agronomically important traits. Candidate genes for these traits in pigeonpea have sequence similarity to genes functionally characterized in other plants for flowering time control, seed development and pod dehiscence. Our findings will allow acceleration of genetic gains for key traits to improve yield and sustainability in pigeonpea.
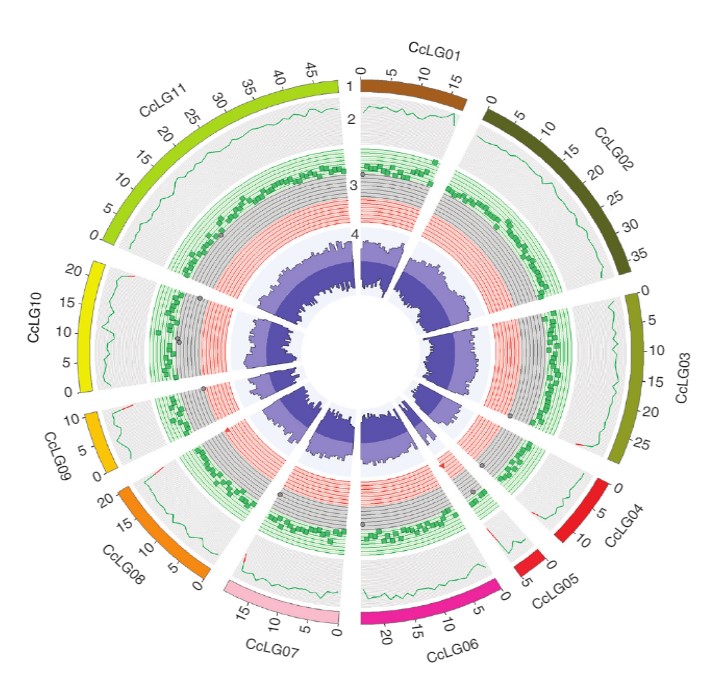
Figure: A Circos image representing variations identified across 292 Cajanus accessions. (1) 11 pseudomolecules (CcLG01 to CcLG11) in different colors with scale of Mb genome. (2) Sequence coverage. The range of the coverage plot axis is 0 to 40,000 (green, >20,000; red, <20,000). (3) Green squares, dark shaded circles and red triangles represent SNP regions in breeding lines, landraces and wild relatives, respectively. The range of the SNP axis is 0 to 36,000 (green square, >24,000; dark shaded circle, 12,000–24,000; red triangle, <12,000). (4) Indels (the range of the indel axis is 0 to 3,500; light purple, insertions; dark purple, deletions).